
More Information
Submitted: 17 December 2019 | Approved: 18 December 2019 | Published: 19 December 2019
How to cite this article: Cárdenas-Moreno Y, Espinosa LA, Vieyto JC, González-Durruthy M, del Monte-Martinez A, et al. Theoretical study on binding interactions of laccase-enzyme from Ganoderma weberianum with multiples ligand substrates with environmental impact. Ann Proteom Bioinform. 2019; 3: 001-009.
DOI: 10.29328/journal.apb.1001007
Copyright License: © 2019 Cárdenas-Moreno Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Laccase; Ganoderma weberianum; Molecular Docking; Bioremediation; Antibiotics; Chlorophenols; Benzidine

Theoretical study on binding interactions of laccase-enzyme from Ganoderma weberianum with multiples ligand substrates with environmental impact
Yosberto Cárdenas-Moreno1*, Luis Ariel Espinosa2, Julio Cesar Vieyto3, Michael González-Durruthy4,5, Alberto del Monte-Martinez6, Gilda Guerra-Rivera1 and Maria Isabel Sánchez López1
1Laboratory of Biotechnology, Department of Microbiology and Virology, Faculty of Biology, University of Havana, Havana, Cuba
2Proteomics Group, Department of System Biology, Center for Genetic Engineering and Biotechnology, P.O. Box 6162 Havana, Cuba
3Computational Biochemistry, Faculty of Biology, Center of Medical Biotechnology, University Duisburg-Essen, Universitätsstr.2, 45117 Essen, Germany
4LAQV-REQUIMTE of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal
5Soft Matter and Molecular Biophysics Group, Department of Applied Physics, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain
6Laboratory of Enzymatic Technology, Center for Protein Studies, Faculty of Biology, University of Havana, Cuba
*Address for Correspondence: Yosberto Cárdenas-Moreno, Laboratory of Biotechnology, Department of Microbiology and Virology, Faculty of Biology, University of Havana, Havana, Cuba, Email: [email protected]
Laccase catalyzes oxidation of lignin and aromatic compound with similar structure to this one. Their low substrate specificity results on degradation of similar phenolic compounds. In this context, Molecular Docking was performed with different ligands suggesting potential bio-degradation. Binding active-sites prediction of fungal laccase (access number uniprotkb: A0A166P2X0), from Ganoderma weberianum was performed using machine learning algorithm based on Deep Convolutional Neural Networks (DeepSite-CNNs chemoinformatic tool). Herein, ligands like 2,4 - dichlorophenol, benzidine, sulfisoxazole, trimethoprim and tetracycline were analyzed and two additional reference controls which were 2,2 – azinobis 3 – ethylbenzothiazoline – 6 - sulfonic acid (ABTS) and 2,6 - dimetoxyphenol (2,6 DMP) were used in comparison with the other former mentioned ligands based on high laccase affinity. The five ligands were carried out because their potential biotechnological interest: the antibiotics sulfisoxazole, trimethoprim and tetracycline, and xenobiotics 2,4 - dichlorophenol and benzidine. Molecular docking experiments returned Gibbs free energy of binding (FEB or affinity) for laccase-ligand complexes. The best docking binding-interaction from each laccase-ligand conformation complexes suggest great ability of these ligands to interact with the laccase active-binding site. Herein, FEB values (kcal/mol) were obtained with higher affinity values for reference controls like 2,6 - dimethoxyphenol with -4.8 Kcal/mol and ABTS with -7.1 Kcal/mol. Furthermore, the FEB values were -4.7, -6.5, -6.8, -5.2 and -6.5 Kcal/mol, for 2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline and trimethoprim respectively with high prevalence of hydrophobic interaction with functional laccase binding residues. Lastly, this study presents for first time at the bioinformatics field a molecular docking approach for the prediction of potential substrate of laccase from Ganoderma weberianum towards biotechnological application.
Laccases (benzenediol: oxygen oxidoreductase, EC 1.10.3.2), that belong to the multicopper, are generally glycoproteins. They have been found in a wide variety of higher plants, fungi, bacteria and insects [1]. Wide range of laccases play different functions depending on the organism and environmental condition. Fungal laccases have been related mainly with pigment production, morphogenesis, sporulation and pathogenesis on plants and animals [2]. Fungi like White-rot fungi releases laccases out of the cell playing an essential roll on the lignocellulose material degradation, contributing to the carbon cycle producing several isoenzymes in some fungal species. Microbial laccases participate in the oxidation of antibiotics such as flavonoids and phytoalexins [3]. Usually these extracellular glycoproteins are monomeric. Their molecular weight varies between 50 and 140 KDa. Most of fungal laccases have a content of 520-550 amino acids, without including signal peptide [4]. Because the complex lignin structure, these enzymes have a low substrate specificity which allow the efficient degradation of this structure. They are able to catalyze the oxidation of a broad range of phenolic substrates, and non-phenolic compound substances in the presence of small mediator molecules, such as (ABTS) and 1-hydroxybenzotriazole (HBT) [5]. Non phenolic substrates, such as ortho and para-diphenols, methoxy-substituted phenols, aromatic amines, phenolic acids and other compounds can be oxidized, coupled to the reduction of molecular oxygen to water with one electron oxidation mechanism [6]. However, it has been proved that it is not required a redox mediator to degrade efficiently simulated dyes affluent by using recombinant laccase from Ganoderma sp [7]. It uses Oxidation reactions in pharmaceutical, chemical and food processing industries and for the wastewater treatment.
Many types of antibiotic act on important physiological or metabolic functions of the bacterial cell [8]. Several microorganisms have developed resistant mechanisms for each one of these antibiotics. In this sense millions of kilograms of antimicrobials are used globally each year in the prophylaxis and treatment of people, animals and agriculture. It finds more and more antimicrobials in wastewaters, increasing remarkably the selection of antibiotic-resistant microorganisms in the environment [9]. Tetracyclines, sulfonamides and trimethoprim represent one of the major amounts of antibiotics in final effluents of wastewaters treatment plants [10]. In this sense laccases have been studied in tetracycline antibiotics degradation [11]. Elimination of carbamazepine by laccases in the presence of mediators has been studied as well [12]. Another experimental study demonstrated the degradation of sulfonamide, tetracycline, and quinolone antibiotics by laccase-mediated oxidation coupled with soil adsorption [13]. Antibiotics norfloxacin and ciprofloxacin degradation by white-rot fungus laccases have been experimentally studied in Trametes versicolor [14]. Phenol oxidases (laccases and tyrosinases) and peroxidases have been established to eliminate the endocrine disrupting chemicals (EDCs) [15].
Xenobiotics like chlorophenols, are synthetics organic compounds extensively used as pesticides, dyes or biocides synthesis. They commonly appear in industrial wastes as direct pollutants. One of them is 2,4 - dichlorophenol (2,4 - DCP), with high impact in aquatic environment [16]. The compound 2,4 - DCP does not mean generally a final commercial product but is an important intermediate that is released into the environment as intermediate compound from several industries; that´s why it is a main pollutant in the aquatic environment in the United States of America as well as in China because of the high toxicity for aquatic life, resistance of degradation, and potential bioaccumulation [17-19] and was the most abundant phenolic compound found in the river water in UK [16]. 2,4 - DCP has been reported as endocrine disruptor [20], causing permanent impairment of vision or blindness of eyes and injury in respiratory tract as human and animals were exposed to it [21]. Studies shown that high concentrations of 2,4 - CP in treated mice induce a significant percentage of chromosome aberrations and sperm-head [22]. On the other hand, benzidine is a synthetic compound used in benzidine dyes production for cloth, paper, and leather and would be present in wastewater from these industries. This compound increases the risk of developing cancer of urinary bladder in people [23]. In addition, The U.S. Department of Health and Human Services (DHHS), the International Agency for Research on Cancer and the US EPA have determined that benzidine is a human carcinogen as well [24]. Immobilized laccase from white-rot fungi Trametes versicolor have been promising an enzymatic remediation of water polluted by benzidine based dyes (i.e., Direct Blue-1 and Direct Red 128) [25]. During the biodegradation of Direct Blue-6, there is induction of oxidative enzymes (Lip, laccase) and tyrosinase [26].
White-rot basidiomycetes´s laccases are great biotechnological candidates on biodegradation of several compounds. Ganoderma sp belongs to this group and their laccases are promising alternative for current treatment technologies. Studies of these laccases have demonstrated efficient degradation compounds like dyes [7]. Nevertheless, none bioinformatic study about antibiotics or other relevant xenobiotic compound for environmental biotechnology, have been reported for Ganoderma weberianum laccases. The implementation of chemo-informatic tools based on Molecular Docking Simulation (MDS) appears to be an efficient strategy for the prediction of ligand-protein interactions of laccase-enzyme (protein) with several substrates (ligand) [27]. The use of MDS is a powerful new platform for the rational design of new drugs (ligands) before its massive production, allowing the computational interaction analysis of a large volume and versatility of designs of agonist, antagonist, specific inhibitors and other substrates with receptors (or key molecular residues of a specific targets) [28-30]. In MDS simulations, hundreds of thousands of orientations and conformations of a protein-ligand inside an enzyme active binding site are evaluated and ranked according to their complex-stability in terms of the estimated free energy of binding (FEB). Besides, Autodock Vina provides functionalities related to the incorporation of receptor flexibility through flexible side chains and ensemble docking [31] in order to improve the conclusions about the in silico results. Docking simulations are not simple due to several entropic and enthalpy factor that influence the receptor-ligands interaction. On the other hand, most of docking tools treat ligands as flexible, but the receptors are treated as rigid and in some cases, only some side chains can be set as flexible or key amino acid residues involved in the enzyme catalysis. Conversely, proteins are biologically dynamic molecules and its flexibility properties are frequently vital to determine their molecular mechanisms and ligand recognition. There is a number of alternative approaches to incorporate the receptor flexibility into docking simulations.
The present study describes the interaction among five compounds with current importance in environmental biotechnology and Ganoderma weberianum laccase (access number uniprotkb: A0A166P2X0). For first time it is used Molecular Docking experiments to understand interactions between laccase form this particular white rot fungi, G. weberianum and relevant chemical compounds which impact the environment, for future applications though environmental biotechnology. The results of this study may be useful in theoretical analyze and prediction of potential compounds (described before) and some others substrates for fungal laccases, specifically Ganoderma weberianum laccase, for future experimental studies on industrial wastewaters treatment.
Performed molecular docking simulation
Molecular docking simulation was carried out to evaluate the integration between laccase-enzyme from Ganoderma weberianum (access number uniprotkb: A0A166P2X0) [32], and several type of ligand laccase-substrates (2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS and 2,6 - dimethoxyphenol), molecular docking simulation. As first step Phyre2 Protein Fold Recognition Server as PDB x-ray structures model [33] was used to modelling the laccase-enzyme structure-files (receptor). The optimization of laccase-enzyme structures was performed by using the AutoDock Tools 4 software for AutoDock Vina software previous to docking experiments. Considering the appropriate hybridization geometry, those hydrogen atoms based on built-in modules were added as a partial charge and protonation states followed by a bond orders assignment and set up rotatable bonds of the laccase-enzyme structures.pdb x-ray structures [31].
Laccase-enzyme ligand obtaining
In the second step, the different types of laccase-enzyme ligand structures, were obtained from Pubchem Data Base Chemical Structure Search as 2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS and 2,6 - dimethoxyphenol [34].
Optimization of Laccase-enzyme ligand structures
The optimization of the laccase-enzyme ligand structures was developed through the MOPAC extension for geometry optimization based on the AM1 - Hamiltonian method [31,35,36].
Evaluation of Laccase-ligand complexes interactions
The Autodock Vina flexible molecular docking, which is an open software source developed by Trott and Olson (2010) [31], was establish to evaluate the complexes or laccase-ligand interactions (free energy of binding or affinity, FEB in Kcal/mol). To analyze the knowledge-based potential and the empirical information, it has been implemented ΔG scoring function. The empirical information has been obtained from experimental binding affinity measurement with Autodock Vina scoring energy functions with Amber force-field parameters [31,37,38]. The flexible docking option favors an enthalpy gain of laccase-ligand complexes non-associated to ligand intra-molecular deformation or vibrational decrease within laccase-active sites. Through Deep Site algorithm [39], laccase-binding active sites were previously predicted, where laccase cavities are identified and delimited, potentially, at the Van der Waals surface able to bind small ligand like 2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS and 2,6 - dimethoxyphenol. This is a machine learning approach, which considers all the molecular descriptor related to protein (laccase-enzyme) and is based on convolutional neural networks (DCNNs). DCNNs is an algorithm able to predict ligand-binding sites provided by a library with an extensive test set with more than 7000 proteins of the scPDB database [40], that validated this Deep Site algorithm [39]. The laccase binding-pocket predictions as well as the laccase-volumetric map prediction were used to establish the cartesian coordinates of docking box simulation like laccase-grid box size, with dimensions of X = 30 Å, Y = 32 Å, Z = 34 Å and the laccase- grid box center X = 14.571 Å, Y = 9.739 Å, Z = 36.997 Å.
Several runs were carried out taking account random conformations and the number of iterations in a run. The former mentioned operation has been implemented by using exhaustiveness option set to 100 (average accuracy) in each docking calculation [31].
The ΔGbind values defines FEB to all docked poses of complexes (laccase-ligands). It includes also the internal steric energy of ligands such as 2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS and 2,6 - dimethoxyphenol. ΔGbind expression is reflected as the sum of individual molecular mechanics terms of standard-chemical potentials like: Van der Waals describing interactions (ΔGvdW), hydrogen bond (ΔGH-bond), such as: electrostatic interactions (ΔGelectrost), hydrogen bond (ΔGH-bond), Van der Waals (ΔGvdW), and intra-molecular ligands interactions (ΔGinternal), obtained and validated from empirically validated Autodock Vina scoring function based on default Amber force-field parameters [31,38].
Besides, Autodock Vina scoring function takes account the optimal-linear FEB coefficients from determined chemical potentials (ΔGinternal). The general thermodynamic equations represented below Equation (1) describes the distance-dependent atom-pair interactions (dij) that determines the overall docking force field parameters.
(Eq. 1)
(Eq. 2)
ΔG = - RT (ln Ki), R (gas constant) is 1.98 cal*(mol*K)-1, and Ki represents the predicted inhibition constants at T = 298.15 K. The Van der Waals interaction as Aij/dij12 and Bij/dij6 (repulsive or hyparabolic function) is represented through the first term of a 12-6/Lennard-Jones potential, which describes a typical Lennard-Jones interaction (laccase-ligands). In the equation dij is the surface distance calculated as dij = rij -Ri - Rj, where rij is the interatomic distance and Ri and Rj are the radio of the atoms in the pair of interaction of laccase(i) - ligands(j) atoms. It also shows the Gaussian term, which is negative, and the parabolic positive. The second term consists of a pair of H-bonds (one donor and one acceptor) as a directional 12-10 hydrogen-bonding potential term such as Bij/dij12 and Cij/dij10, where E (t) is a very important element that represents the directionality of the hydrogen bonds and dij follows the criteria mentioned above. The third term describes the Coulomb electrostatic potential in the formed complex (laccase-ligands) of N charges (qi, qj) of pairs of charged atoms of laccase (i) and ligands (j). It assigned in this case Gasteiger partial atomic charges of the laccase-enzyme. Herein, dij is the interatomic distance between the point charges as the reference positions of interaction based on distance-dependent dielectric constant. To validate the internal steric energy of each laccase-ligand including dispersion-repulsion energy and a torsional energy it used (ΔGinternal) (the fourth term of the equation (3). This was obtained through the sum of the default Amber force field parameters (ligand conformation-independent parameters of the Autodock Vina scoring function) [31]. It added the electrostatic components. Polar and non-polar hydrogen atoms were assigned and SWCNT-partial atomic charges through the Gasteiger-Huckel algorithm [41,42] and before the using of a partial equalization of orbital electro negativities (PEOE) method for charges. To determine the Molecular docking dimensionality, based on the degree of freedom (DOF) of each component of the laccase-ligand data set (2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS, 2,6 - dimethoxyphenol) it had in consideration these components: ligand-number of rotatable bonds/torsion (tor1, tor2,…, torn = Ntor), ligand-atom orientation/quaternion (q(xi), q(yi), q(zi), q(wi) = 4) and ligand-atom position/translation (xi, yi, zi = 3). It does not have a remarkable weight on the FEBdock the Ligand-total dimensionality (total DOF = 3 + 4 + n) due to the very small intra-molecular contributions of force field parameters of the laccase-ligand. These are considered as rigid and taking account the ligand-geometry optimization (described above) [43] based on the ΔGinternal minimization of all the laccase-ligands used in the present study.
It was obtained Laccase-ligand conformations with the lowest Gibbs docking free energy of binding (FEB negatives value). To select the best root-mean-square deviation (RMSD). It was taken as a criterion of correct docking pose accuracy below 2 Å according to the equation [5].
(Eq. 3)
Finally, it analyzed the results obtained from the molecular docking taking account the final free energy of binding (FEB values) (for/to) the laccase-ligand complexes of each docking experiment. As reference controls on laccase active-binding pockets it analyzed and used the 2,6 DMP-ligand molecule and ABTS-ligand.
An important task to ensure accuracy of our theoretical data consists in the prediction of feasible laccase-binding sites. Several methods for detecting enzyme-binding cavities have been developed over the years based on structural, geometric, and chemical features of the protein (laccase). In the present study the prediction of binding active-sites of laccase from Ganoderma weberianum was performed using machine learning algorithm based deep convolutional neural networks (Deep Site-CNNs chemoinformatic tool) which was previously validated by providing an extensive test set based on more than 7000 proteins of the scPDB database. The results on prediction/identification of the laccase binding-site are shown in figures 1,2.
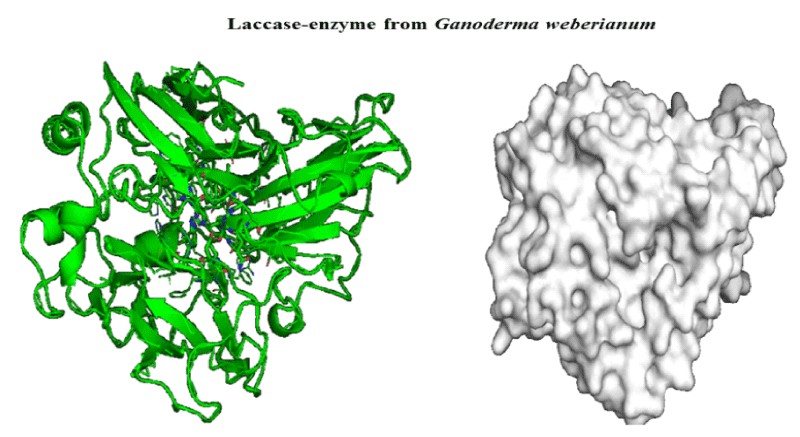
Figure 1: 3D-structure of laccase-enzyme from Ganoderma weberianum (access number uniprotkb: A0A166P2X0).
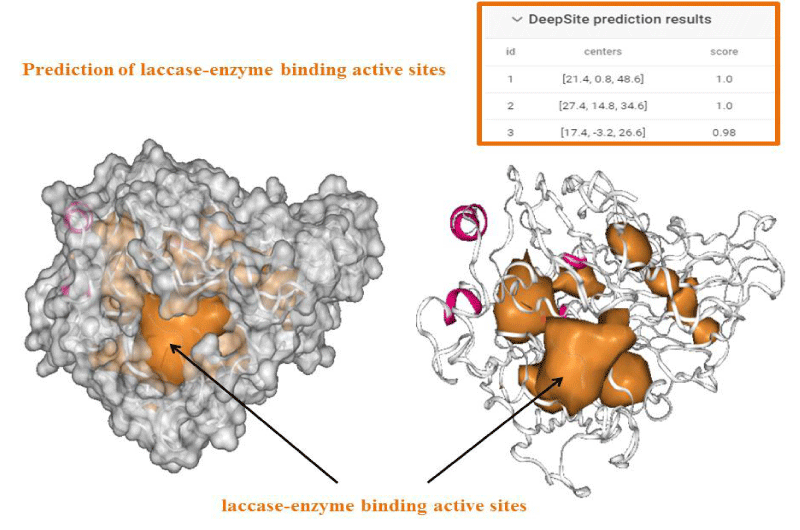
Figure 2: Deep Site prediction of binding active-sites of laccase from Ganoderma weberianum (access number uniprotkb: A0A166P2X0).
Herein, through the Ramachandran diagram which is a 2D - projection on the plane from laccase 3D - structure are depicted all the possible conformations of each laccase-residue including the laccase-active binding sites defined by dihedral angles (Psi) and (Phi) around the peptide-bond of the laccase-residues [44]. In this case, within the bordering lines of the Ramachandran diagram (conformational favored residues) is found the allowed torsion values of Psi vs Phi of a given laccase-residue. On the other hand, is considered as disallowed the torsion values of dihedral angles Psi vs Phi appeared outside the Ramachandran bordering lines, which are conformational non-favored residues. Herein, note that the laccase-enzyme.pdb model obtained by Phyre2 does not present disallowed residues-based Ramachandran outlier; except the Glycine-laccase residues, which is not considering as active binding site residues. Then, the absence of false positives on flexible-docking interactions was corroborated for all molecular docking experiments (Figure 3).
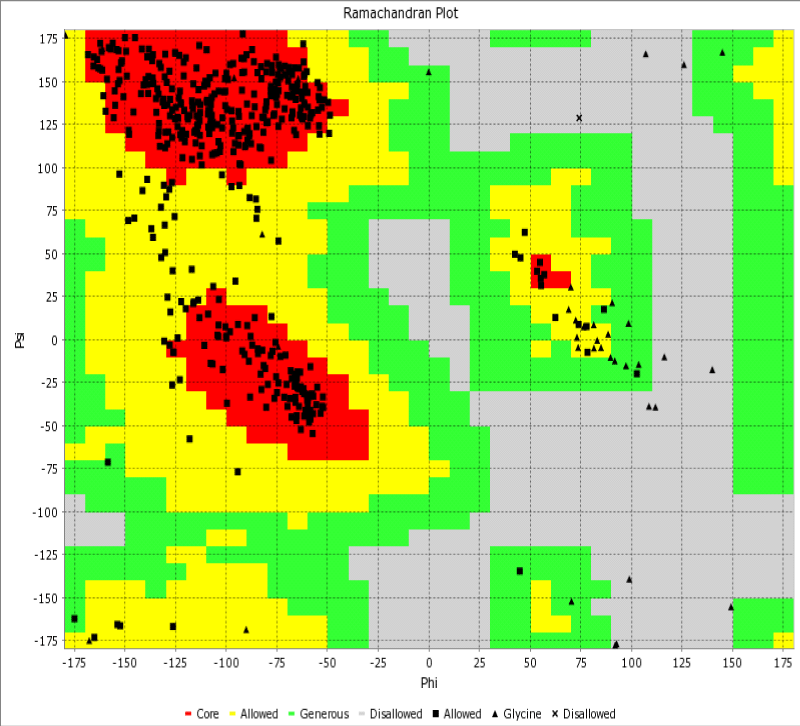
Figure 3: Representation of Ramachandran diagrams and spatial distribution of Ramachandran outliers (Glycine) in the modeled laccase.pdb x-ray structure analyzed (right). Figure shows all the possible combinations of dihedral angles of torsion Psi versus Phi of each amino acid residues of laccase-enzyme modeled.
Then, we carried out the molecular docking experiments to obtain the Gibbs free energy of binding (FEB or affinity) for the complexes formed between laccase-enzyme and the different ligands. Docking results either are considered as energetically unfavorable when Gibbs free energy of binding for laccase-ligand-substrates complex ≥ 0 Kcal/mol pointing extremely low or complete absence of affinity. Herein, the general results of molecular docking are shown in the table 1.
| Table 1: Results of docking complexes of interaction for the best laccase-ligand conformations based on the affinity in Kcal/mol. | |||
| Complexes | Ligand structure | Docking Affinity (FEB: Kcal\mol) | R.M.S.D (Å) |
| 2,4-dichlorophenol-laccase | 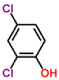 |
-4.7 | 1.432 |
| Benzidine-laccase | 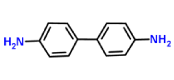 |
-6.5 | 0.245 |
| Sulfisoxazole-laccase | 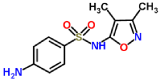 |
-6.8 | 1.991 |
| Tetracycline-laccase | 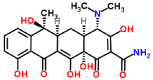 |
-5.2 | 1.318 |
| Trimethoprim-laccase | 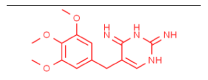 |
-6.5 | 0.096 |
|
ABTS-laccase (control 1) |
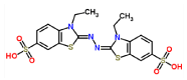 |
-7.1 | 3.487 |
|
2,6-dimethoxyphenol-laccase (control 2) |
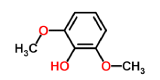 |
-4.8 | 2.159 |
According to the obtained results, a mechanistic interpretation from the best laccase-ligand conformations based on the affinity in Kcal/mol allowed to expand understanding the interaction, amino acids residues involved as a better characterization of these interactions. The best docking binding-interaction (RMSD < 2 Å) from each laccase-ligand conformation complexes suggest great ability of these substrates to interacting with the laccase active-binding site involved in oxidation of phenolic or some non-phenolics compounds. The strength of interactions based on the FEB-values of the laccase-ligand conformation complexes have the following order:
FEB (ABTS-laccase) > FEB (sulfisoxazole-laccase) > FEB (trimethoprim-laccase) ~ FEB (benzidine-laccase) > FEB (tetracycline-laccase) > FEB (2,6 - dimethoxyphenol-laccase) > FEB (2,4 - dichlorophenol-laccase). With relevant interatomic distance of interaction lower than 7 Å in all the cases (da Silveira, et al, 2009).
The FEB (ABTS-laccase complex) value used as control experiment (control 1) was the most negative interaction energy (strong interaction) compared with the remaining laccase-ligand conformation complexes maybe due to the high number of hydrophobic interactions associated to non-ligand laccase-residues (Pro 415, Thr 451, Pro 452, Ile 476, Phe 183, Leu 185, Phe 286, Asn 285). Also, it is important to note, the presence of interactions as polarized and electrostatic hydrogen-bonds of small and short barrier according to the Euclidean interatomic distances values (dij: ligand-laccase interatomic distance < 7 Å) between oxygen atoms of ABTS and nitrogen atoms of laccase residues (Ala 453, His 479) (Figure 4).
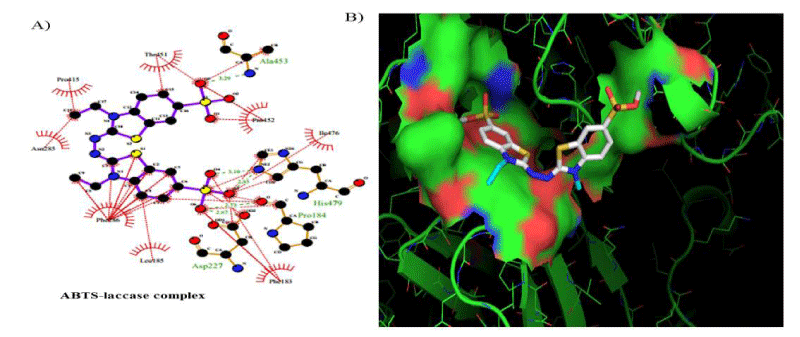
Figure 4: 2D Lig-Plot diagram (A) and Van-der-Waals surface of laccase active-site representation. (B) for the ABTS-laccase interactions used as control experiment 1.
However, the strength of 2,6-dimethoxyphenol-laccase interactions used as control experiment (control 2) showed to be relatively low compared with the remaining laccase-ligand conformation complexes and similar to the 2.4 - diclorophenol-laccase complex due to smaller number of hydrophobic interactions associated to non-ligand laccase-residues like Asp227, Pro184, Phe183, Ile476, Ala414, Asn285, Phe286, N-(Ɛ)-atom and C-(Ɛ)-atoms-His 479 and the oxygen - (1) of 2,6 - dimethoxyphenol. Also considering a single interaction hydrogen-bond between N- N-(Ɛ)-atom-His 479 and the oxygen-(1)-atom of the 2,6 - dimethoxyphenol. The docking hydrogen bonds in the most of cases tested are linear docking interaction, which minimizing the repulsion between partial negative charges of the electronegative oxygen atoms with Euclidean distance of 3.12 Å. In this sense, it is important to note the presence of inter-atomic repulsion between oxygen atoms from 2,6 - dimethoxyphenol and oxygen atoms in the laccase-active site (Figure 5).
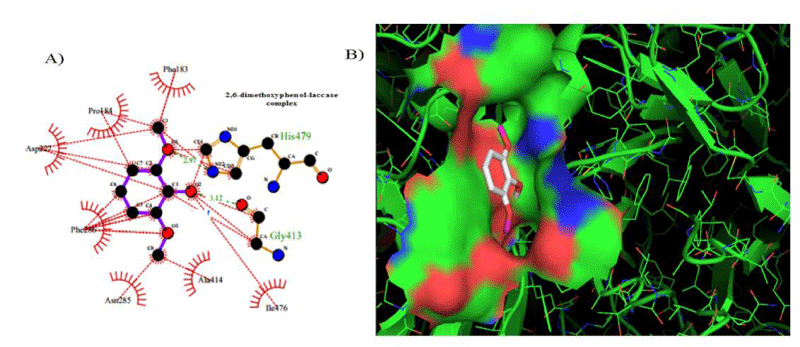
Figure 5: 2D Lig-Plot diagram (A) and Van der Waals surface of laccase active-site representation (B) for the 2,6-DMP laccase interactions used as control experiment 2.
Furthermore, best docking crystallographic binding position for the remaining laccase-ligands (2,4 - dichlorophenol, benzidine, sulfisoxazole, tetracycline, trimethoprim, ABTS, 2,6 - dimethoxyphenol) showed great predominance of specific-hydrophobic interactions and also with different biophysical interaction-environment (different active-site residues) the results are shown in the table 2.
Table 2: Specific-hydrophobic interactions of the best docking crystallographic binding position of the laccase-ligands tested. |
|
| Complexes | Hydrophobic Interactions |
| 2,4-dichlorophenol-laccase | 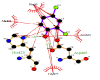 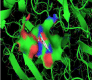 |
| Benzidine-laccase | 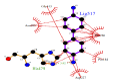 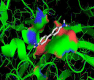 |
| Sulfisoxazole-laccase | 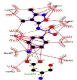  |
| Tetracycline-laccase | 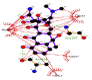  |
| Trimethoprim-laccase |  |
A bioinformatic analyze allows to understand in details, the substrate interaction and amino acid residues involved in the laccase interaction. In addition, MDS allows the better understanding of ligand-protein interaction patterns after docking simulations and calculate their interactions by using an algorithm to verify the amino acid residues in contact with the specific substrates evaluated. The studied compounds were 2,4 - diclorophenol, benzidine and three commercial antibiotics of wide spectrum use: Sulfisoxazole, Tetraciclyne and Trimethoprim. Controls compounds ABTS and 2.6 - dimetoxyphenol were analyzed as well because represent some of the most specific substrates for Ganoderma sp. laccases. We made an analysis of those interactions and amino acids residues involved by using bioinformatics tools such as Molecular Docking Simulation (MDS) with Deep Site program.
The enzymatic properties of fungal laccases vary greatly. The most affinity substrates reported are ABTS and DMP. The Km (µM) ranges vary a lot such as: ABTS from 4-770, DMP from 26-14720 and Kcat (S-1) vary in a broad range as: ABTS from 198-350000 and DMP from 100-360000 [45]. The present results show the best affinity value for ABTS in comparison to the second control (2,6 DMP), in agreement with experimental result afore mentioned. Stronger interaction was observed for ABTS and the major RMSD value firstly due high density of chemical groups, which explain the high number of amino acids residues interacting. Similar numbers of amino acids have been generated as model interaction between recombinant Ganoderma lucidum laccase, recombinant Pleurotus ostreatus and ABTS [4].
The typical four copper-binding conserved domains of laccases were checked: CuI (HWHGFFQ), CuII(HSHLSTQ), CuIII (HPFHLHG) and CuIV (HCHIDFHL) [7,32]. A strongly conserved pattern of four His–X–His motifs is present. Excepting CuII domain, ligands interacted with the resting sites (CuI, CuIII and CuIV): ABTS and 2,6 dimetoxyphenol with CuIV (Ile476 and His479), 2,4 diclorophenol and sulfisoxazole with CuI (Phe90) and CuIII (Gly422, His423 and Ala424) and finally benzidine and trimethoprim with CuIV (His479) motif. Affinity values for controls demonstrated the use of these substrates for laccase, being ABTS the highest. Curiously tetraciclyne has no interaction with any copper binding site and have a higher affinity (-5.2 Kcal/mol) in comparison with others compounds maybe for the high density of polar groups unable to interact with the amino acids residues that conform the active sites Cu. Benzidine, sulfisoxazole, trimethoprim and ABTS were the best affinity values. Complexes 2,4 diclorophenol-lacasse and Sulfisoxazole-laccase didn´t show any common amino acidic residue with used controls or others analyzed ligands. But goods affinity values (-4.7 Kcal/mol for 2,4 diclorophenol) similar to 2,6 DMP (-4.8 Kcal/mol) and goods affinity for Sulfisoxazole-laccase complex (-6.5 Kcal/mol). However, this complex shares common amino acidic residues involved in their interactions: Phe90, Gly 422, His 423, Ala 424, Asp 465 and Asn 466. The Gly422, His 423, Ala 424 sequence are located in a conserved region in fungi that belongs to CuIII binding site.
Complex benzidine-lacases was similar to ABTS-laccases complex with high affinity values of -6.5 Kcal/mol and -7.1 Kcal/mol respectively. However, lower RMSD for benzidine-lacase (0.245 Å) complex because stereo specific structure less complex than ABTS (3.487 Å). Benzidine ligand is a symmetric molecule having two amino groups (-NH2). Each amino is linked to a biphenyl group in position -para. Docking results between this ligand and laccase show high affinity (-6.5 kcal/mol, Table 1) and one possible explication is due to the strong interaction of His479 with benzidine ligand through π-π-stacking and H-bond interactions. Imidazole ring of His residue has aromatic properties that explain the pi-stacking interaction with phenyl group. Interestingly, His479 is part of the highly conserved H-X-H motif that interacts with copper ion during laccase-mediated catalysis. In addition, an H-bond was detected between amino (-NH2) of benzidine and protonated nitrogen (-NH-) of imidazole ring in His479. This strong interaction could explain the small value of RMSD (0.245 Å, Table 1) for benzidine-laccase complex. It is also observed in the case of other ligands, such as trimethoprim and sulfisoxazole. His423 is located in the H-X-H motif in the copper-binding site CuIII and interact exclusively with 2,4 - diclorophenol and sulfisoxazole ligands. H-bond and pi-stacking aromatic interactions were observed between imidazole ring of His and the hydroxyl and phenyl group of 2,4 - diclorophenol ligand. In the case of sulfisoxazole only hydrophobic interaction with His423 was detected. In addition, His479 is near to other H-X-H motif and interacts with ABTS, 2,6 - dimethoxyphenol, benzidine and trimethoprim. In the case of the first two ligands (ABTS and 2,6 DMP), the interaction was through H-bond. The Asp residues are involved in each binding interaction. It would suggest a crucial role in proton abstraction from the substrate, but in plant laccase Asp residue instead of Asp, perform the same role [46-48]. Similar to others reports from Awasthi and collaborators in 2014, in our study Asp227 has been observed at the active site of fungal laccase. Studies with lignin models compounds (sinapyl, coniferyl and p-coumaryl alcohol) and laccase from White rot fungi Phlebia brevispora and Dichomitus squalens showed FEB values between -5,4 and -7,8 respectively. (Table 3).
Commonly found amino acid residues in white rot fungal laccases were Pro, His, Ser, Phe, Gly, Ala, Tyr, Leu, Lys Gln and Thr [28]. Amino acid residue Phe have been found in fungal laccases interaction. In our study, Phe (mainly Phe286) was present in each binding interaction as well. These characteristic is a possible key in the high redox potential of some laccases, because is observed in Trametes versicolor laccase which he substitution instead methionine (in plant laccases) increase the redox potential in comparison with plant laccase [46,49,50] making more efficient on lignin degradation. Awasthi, et al. in 2014 found for fungal laccase from Trametes versicolor 11 residues involved in binding interactions with lignin models compounds: Leu185, Asp227, Asp229, Phe260, Ser285, Phe286, Gly413, Ala414, Pro415, Ile476 and His479. These residues were commonly in the binding interactions with all the model compounds and some residues are represented in our study, excepting Ser that it is not present in any binding interaction.
The present study addresses the docking interactions between laccase and several compounds (2,4 diclorophenol, benzidine, tetraciclyne, trimetroprim with potential relevance in environmental biotechnology. In general terms, the obtained Gibbs free energy values of affinity, pointing that the stabilization of the laccase-ligand complexes are mainly based in non-covalent hydrophobic interactions with relevant inter-atomic distance of interaction lower than 7 Å in all the cases. The binding-affinity obtained from the best laccase-ligand docking complexes following the order FEB (ABTS-laccase) > FEB (sulfisoxazole-laccase) > FEB (trimethoprim-laccase) ~ FEB (benzidine-laccase) > FEB (tetracycline-laccase) > FEB (2,6 – dimethoxyphenol-laccase) > FEB (2,4 - dichlorophenol-laccase). According to this, the controls simulation experiments using ABTS and 2,6 dimethoxyphenol confirm that the free energy of binding FEB of obtained laccase-ligand complexes are mainly based in hydrophobic interactions suggesting an efficient mechanism biodegradation for the compounds studied. Due to the stability of the obtained docking complexes with negatives FEB values in all the cases. Besides, the results on structural modeling revealed that the laccase enzyme can be efficiently modeled with conformationally favored binding-site residues to explain interaction mechanisms of new ligands and or substrates of laccase enzyme with potential environmental impact. Finally, these theoretical evidences open new perspectives toward environmental biotechnology applications through in vitro experiments using G. weberianum laccase and to implement strategies of bioremediation of wastewaters contaminated with similar compounds derived from pharmaceutical or other industries.
The authors thank to Center of Genetic Engineering and Biotechnology and to the National Proyecto of Basic Sciences, from Cuba. We also thank the collaboration of lic. Maria Carla Pi Noroña and Adiza Veronica Ameh.
- Viswanath B, Rajesh B, Janardhan A, Kumar AP, Narasimha G. Fungal Laccases and Their Applications in Bioremediation. Enzyme Res. 2014: 2014: 1-21.
- Baldrian P. Fungal laccases - occurrence and properties. FEMS Microbiol Rev. 2006; 30: 215-242. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16472305
- Cázares-García SV, Vázquez-Garcidueñas MS, Vázquez-Marrufo G. Structural and Phylogenetic Analysis of Laccases from Trichoderma: A Bioinformatic Approach. PLoS One. 2013; 8: 55295. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23383142
- Rivera-Hoyos CM, Morales-Álvarez ED, Poveda-Cuevas SA, Reyes-Guzmán EA, Poutou-Piñales RA, et al. Computational analysis and low-scale constitutive expression of laccases synthetic genes GLLCC1 from Ganoderma lucidum and POXA 1B from Pleurotus ostreatus in pichia pastoris. PLoS One. 2015; 10: 0116524. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25611746
- Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep. 2011; 28: 1883-1896. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21918777
- Latch DE, Packer JL, Stender BL, VanOverbeke J, Arnold WA, et al. Aqueous photochemistry of triclosan: Formation of 2,4 - dichlorophenol, 2,8 - dichlorodibenzo-p-dioxin, and oligomerization products. Environ Toxicol Chem. 2005; 24: 517-525. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15779749
- Zhuo R, Ma L, Fan F, Gong Y, Wan X, et al. Decolorization of different dyes by a newly isolated white-rot fungi strain Ganoderma sp.En3 and cloning and functional analysis of its laccase gene. J Hazard Mater. 2011; 192: 855-873. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21733624
- Philip PR, Lee R, Lin C. The Antibiotic Paradox: How the Misuse of Antibiotics Destroys Their Curative Powers (review). Perspect Biol Med. 2007.
- Kümmerer K. Promoting resistance by the emission of antibiotics from hospitals and households into effluent. Clin Microbiol Infect. 2003; 9: 1203-1214. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14686985
- Miao XS, Bishay F, Chen M, Metcalfe CD. Occurrence of antimicrobials in the final effluents of wastewater treatment plants in Canada. Environ Sci Technol. 2004; 38: 3533-3541. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15296302
- Suda T, Hata T, Kawai S, Okamura H, Nishida T. Treatment of tetracycline antibiotics by laccase in the presence of 1-hydroxybenzotriazole. Bioresour Technol. 2012; 103: 498-501. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22071243
- Hata T, Shintate H, Kawai S, Okamura H, Nishida T. Elimination of carbamazepine by repeated treatment with laccase in the presence of 1-hydroxybenzotriazole. J Hazard Mater. 2010; 181: 1175-1178. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20619797
- Ding H, Wu Y, Zou B, Lou Q, Zhang W, et al. Simultaneous removal and degradation characteristics of sulfonamide, tetracycline, and quinolone antibiotics by laccase-mediated oxidation coupled with soil adsorption. J Hazard Mater. 2016; 307: 350-358. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26826938
- Prieto A, Möder M, Rodil R, Adrian L, Marco-Urrea E. Degradation of the antibiotics norfloxacin and ciprofloxacin by a white-rot fungus and identification of degradation products. Bioresour Technol. 2011; 102: 10987-10995. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21937221
- Macellaro G, Pezzella C, Cicatiello P, Sannia G, Piscitelli A. Fungal Laccases Degradation of Endocrine Disrupting Compounds. Biomed Res Int. 2014; 2014: 614038. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24829908
- House WA, Leach D, Long JLA, Cranwell P, Smith C, et al. Micro-organic compounds in the Humber rivers. Sci Total Environ. 1997.
- Gao J, Liu L, Liu X, Zhou H, Huang S, et al. Levels and spatial distribution of chlorophenols - 2,4-Dichlorophenol, 2,4,6-trichlorophenol, and pentachlorophenol in surface water of China. Chemosphere. 2008; 71: 1181-1187. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18037470
- Jin X, Zha J, Xu Y, Wang Z, Kumaran SS. Derivation of aquatic predicted no-effect concentration (PNEC) for 2,4-dichlorophenol: Comparing native species data with non-native species data. Chemosphere. 2011; 84: 1506-1511. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21543105
- Yin D, Jin H, Yu L, Hu S. Deriving freshwater quality criteria for 2,4-dichlorophenol for protection of aquatic life in China. Environ Pollut. 2003; 122: 217-222. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12531309
- Bhadauria V, Zhao WS, Wang LX, Zhang Y, Liu JH, et al. Advances in fungal proteomics. Microbiol Res. 2007.
- Chemical Advisory and Notice of Potential Risk: Skin exposure to molten 2,4 - Dichlorophenol can Cause Rapid Death, USEPA.
- Amer SM, Aly FAE. Genotoxic effect of 2,4 - dichlorophenoxy acetic acid and its metabolite 2,4 - dichlorophenol in mouse. Mutat. Res. 2001; 494: 1-12. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11423340
- Agency for Toxic Substances and Disease Registry. 2019.
- Choudhary G. Human health perspectives on environmental exposure to benzidine: A review. Chemosphere. 1996. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8581430
- Karagoz B, Bayramoglu G, Altintas B, Bicak N, Yakup MA. Amine functional monodisperse microbeads via precipitation polymerization of N-vinyl formamide: Immobilized laccase for benzidine based dyes degradation. Bioresour Technol. 2011. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21531131
- Kalme SD, Parshetti GK, Jadhav SU, Govindwar SP. Biodegradation of benzidine based dye Direct Blue-6 by Pseudomonas desmolyticum NCIM 2112. Bioresour Technol. 2007. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16822666
- Martínez-Sotres C, Rutiaga-Quiñones JG, Herrera-Bucio R, Gallo M, López-Albarrán P. Molecular docking insights into the inhibition of laccase activity by medicarpin. Wood Sci Technol. 2015.
- Kameshwar AKS, Barber R, Qin W. Comparative modeling and molecular docking analysis of white, brown and soft rot fungal laccases using lignin model compounds for understanding the structural and functional properties of laccases. J Mol Graph Model. 2018. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29127854
- Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA, et al. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016; 116: 6370-6390. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27074285
- Zsila F, Bikadi Z, Malik D, Hari P, Pechan I, et al. Evaluation of drug-human serum albumin binding interactions with support vector machine aided online automated docking. Bioinformatics. 2011; 27: 1806-1813. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21593135
- Trott O, Olson AJ. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31: 455-461.
- Zhou YP, Chen QH, Xiao YN, Ke DS, Tian CE. Gene cloning and characterization of a novel laccase from the tropical white-rot fungus Ganoderma weberianum TZC-1. Appl Biochem Microbiol. 2014.
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015; 10: 845-858. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25950237
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016; 44: 1202-1213.
- McNamara JP, Hillier IH. Semi-empirical molecular orbital methods including dispersion corrections for the accurate prediction of the full range of intermolecular interactions in biomolecules. Phys Chem Chem Phys. 2007; 9: 2362-2370. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17492099
- Govender K, Gao J, Naidoo KJ. AM1/d-CB1: A semiempirical model for QM/MM simulations of chemical glycobiology systems. J Chem Theory Comput. 2014; 10: 4694-4707. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26120288
- Liu J, Wang R. Classification of current scoring functions. J Chem Inf Model. 2015; 55: 475-482. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25647463
- Quiroga R, Villarreal MA. Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS One. 2016; 11. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27171006
- Jiménez J, Doerr S, Martínez-Rosell G, Rose AS, De Fabritiis G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics. 2017; 33: 3036-3042. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28575181
- Desaphy J, Bret G, Rognan D, Kellenberger E. Sc-PDB: A 3D-database of ligandable binding sites-10 years on. Nucleic Acids Res. 2015; 43: 399-404. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25300483
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron. 1980.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009; 30: 2785-2791. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19399780
- González-Durruthy M, Werhli AV, Seus V, Machado KS, et al, Decrypting Strong and Weak Single-Walled Carbon Nanotubes Interactions with Mitochondrial Voltage-Dependent Anion Channels Using Molecular Docking and Perturbation Theory. Sci Rep. 2017; 7: 13271. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29038520
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010; 66: 12-21. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20057044
- Weirick T, Sahu SS, Mahalingam R, Kaundal R. LacSubPred: Predicting subtypes of Laccases, an important lignin metabolism-related enzyme class, using in silico approaches, BMC Bioinformatics. 2014; 15: 15. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25350584
- Awasthi M, Jaiswal N, Singh S, Pandey VP, Dwivedi UN. Molecular docking and dynamics simulation analyses unraveling the differential enzymatic catalysis by plant and fungal laccases with respect to lignin biosynthesis and degradation. J Biomol Struct Dyn. 2015; 33: 1835-1849. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25301391
- Frasconi M, Favero G, Boer H, Koivula A, Mazzei F. Kinetic and biochemical properties of high and low redox potential laccases from fungal and plant origin. Biochim Biophys Acta - Proteins Proteomics. 2010; 1804: 899-908. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20056172
- Madzak C, Mimmi MC, Caminade E, Brault A, Baumberger S, et al. Shifting the optimal pH of activity for a laccase from the fungus Trametes versicolor by structure-based mutagenesis. Protein Eng Des Sel. 2006; 19: 77-84. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16368720
- Gorbacheva MA, Shumakovich GP, Morozova OV, Zaitseva EA, Shleev SV. Comparative Study of Biocatalytic Reactions of High and Low Redox Potential Fungal and Plant Laccases. Vestn Mosk Univ. 2008.
- Madhavi V, Lele SS. Laccase properties, use. Bio Resources. 2009; 4: 1694-1717.